Regulatory Landscape and the Evolution of Contamination Control in 2025
In a recent ATMP article, we went in-depth on the origin of regulatory-related manufacturing challenges seen in the industry and what guidelines need to be applied early on. Now, we examine the process production risks associated with certain ATMP products and how we might address them with an effective contamination control strategy.
Many challenges have been met in ATMP manufacturing (Table 1), of which the development of a proper Contamination Control Strategy (CCS) for ATMPs is a frequently recurring one. The lack of a proper CCS may lead to recalls, regulatory scrutiny, and (temporary) revoking of manufacturing licenses. The CCS requires identifying, scientifically evaluating, and controlling potential risks related to product quality and patient safety. Progress has previously written in detail about CCS (see Progress: Notes on Annex-1).
Contamination Control Strategy and Ph. Eur. Updates
One part of CCS for ATMPs is the prevention and monitoring of bioburden levels. High bioburden levels may accelerate drug product degradation and inevitably affect drug potency. In the case of a contaminated product, a facility may be shut down for extended periods until the source is identified and removed.
Many smaller drugs can be manufactured sterile by incorporating viral or bacterial removal steps after critical manufacturing stages to ensure low in-process bioburden levels. These steps typically enable high drug purity (i.e., fewer contaminants in the case of ATMPs), which supports a more consistent drug potency level. However, in-process removal of contaminants for ATMPs is challenging because ATMPs tend to be larger, unstable, and prone to aggregation (often clogging filters), and share physiochemical properties with potential bacterial or viral contaminants.
Additionally, detecting contamination is more complex due to the similar biophysical properties of ATMPs and potential impurities, which complicates the use of standard industry assays. As a result, more sensitive and product-specific analytical methods are increasingly necessary in 2025. The updated European Pharmacopoeia (Ph. Eur.) further reinforces this by emphasizing flexibility in testing: General Chapter 5.34 and Monograph 3186, implemented in 2025, now support a risk-based approach. For example, manufacturers may use droplet digital PCR (ddPCR) instead of traditional qPCR for impurity testing, and replication-competent virus (RCV) testing may be omitted from final lots if adequately performed at earlier stages. Importantly, the new framework does not impose numerical limits but requires scientific justification and demonstration of impurity clearance.
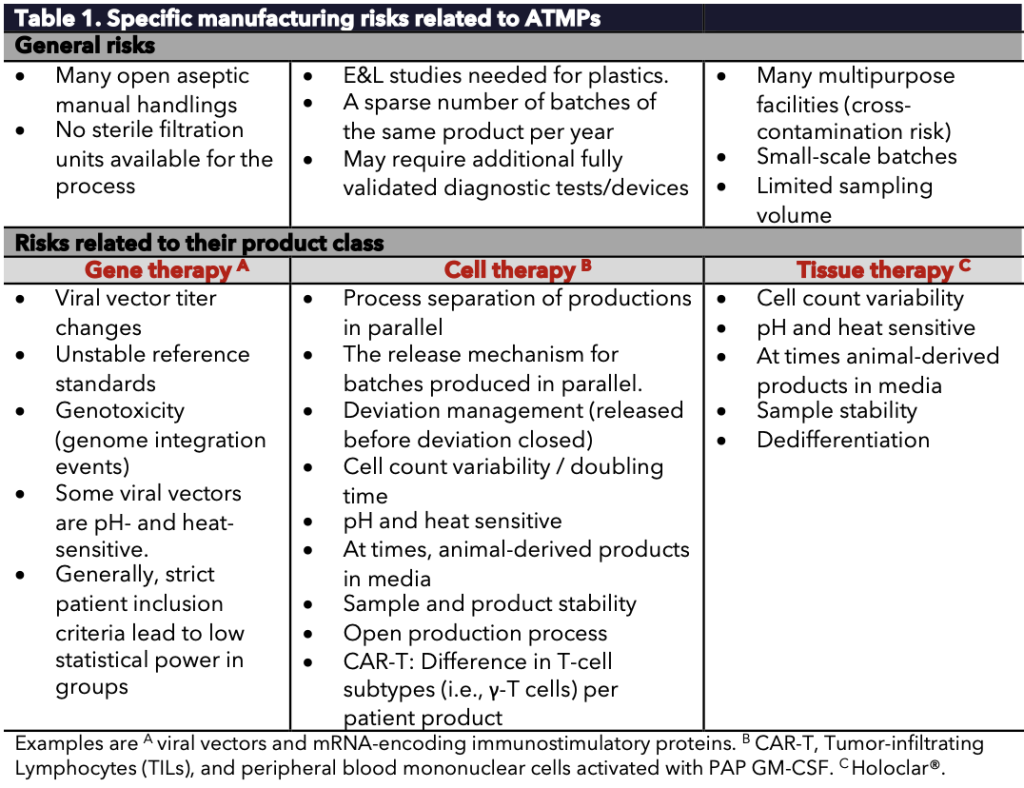
Table 1. Specific manufacturing risks related to ATMPs.
Single-use connectors and Closed Systems
Single-use sterile connectors and high cleanroom-grade areas are common strategies to reduce contamination risks in the ATMP field. For example, working in a cleanroom Grade B instead of D can reduce the bioburden risk by >95%.
We also see that many Grade A isolators in Grade C/D cleanrooms are increasingly installed instead of the more traditional BSCs in Grade A/B cleanrooms. These Restricted Access Barrier Systems (RABS) improve aseptic performance and process separation for ATMP processes.
Tube welding or sterile connectors are the preferred choices for many ATMP processes. The closed tubes or pre-assembled tube kits are γ-irradiated, autoclaved, and packed. Then, the tubes are welded in the manufacturing rooms where every connection is visually inspected.
However, tube welding may introduce particles, complicating validation studies. Additionally, single-use plastic connections may require more extractables and leachables (E\&L) studies.
Yet, a closed process (e.g., by tube-welding and sterile connectors) can be operated in a ballroom Grade C cleanroom, enabling the production of multiple products side-by-side instead of maintaining many small product- or campaign-specific Grade B cleanrooms.
This strategy has gained further traction with the introduction of Annex 1 (2023), which explicitly encourages closed systems and risk-based environmental classification.
Yields and Gene Therapy Standards
Another ATMP manufacturing risk is variability in product yield, the purity of starting material, and the (genetic) stability of the product. For example, differences in (donor) starting material for cell therapy products increase cell count variability and affect batch success rates. Patient-specific factors—such as genetic background or chemotherapy history—can influence outcomes.
Many hospital techniques—such as needle-puncture biopsies or apheresis—may introduce microorganisms from skin surfaces or glands into ATMP starting materials. This risk is particularly high in autologous therapies, where patient-derived inputs make standardization difficult.
This is a much smaller challenge for viral vector gene therapies, which typically use well-characterized, germ-free cell lines as starting materials. However, these therapies face other challenges, including (epi)genetic drifts that may alter viral vector titers over time, unstable reference standards, and complex potency assays. These assays must demonstrate the consistent biological activity of a transgene introduced to a target cell.
The revised Ph. Eur. now provides more refined guidance here, including updates to critical quality testing methods:
- Flow cytometry (2.7.24) for viability and cell count
- BET using recombinant Factor C (2.6.14)
- Microbiological examination of cell-based preparations (2.6.27)
- Nucleated cell viability (2.7.29)
These methodologies support precision testing and product consistency, particularly for genetically modified cell-based therapies.
Progress’ Role in Regulatory Advancement
To support the safe and effective translation of new gene therapy technologies to the clinic, Progress is actively contributing to regulatory development, particularly in the context of gene editing and non-viral delivery platforms. In collaboration with Regulatory Science Network Netherlands (RSNN), Progress is involved in a multistakeholder expert work group focused on therapeutic in vivo gene editing. This initiative, carried out with experts Lourens Bloem and Renske ten Ham, explores how current regulatory frameworks (CCMO, CBG/EMA) can be improved to better accommodate innovations such as CRISPR and platform-based gene editing tools. The goal is to avoid regulatory bottlenecks that hinder the accessibility of advanced therapies. Given the limited existing guidance, the timing is right to evaluate whether current frameworks are still fit-for-purpose. The initiative has national visibility and has attracted interest from the Committee for Advanced Therapies (CAT) of the EMA, which has expressed interest in participating. Thilo Buck represents Progress in this endeavor.
Conclusion
In summary, ATMPs have a higher risk for contamination and require a tighter contamination control strategy and risk reduction steps than traditional small-molecule or monoclonal antibody products. In 2025, CCS is no longer a regulatory recommendation—it is a regulatory expectation, with inspectors focusing heavily on its integration throughout the product lifecycle, not only in aseptic processing but across facility design, raw material handling, personnel behavior, and process validation.
At the same time, evolving frameworks such as Annex 1 and the revised Ph. Eur. chapters show that science-based flexibility, advanced analytics, and proactive regulatory participation are key to the successful and compliant production of gene therapy products.